There is no approved disease-modifying therapy for May-Hegglin anomaly (MHA), and therapeutics focuses on symptom management. Targeted therapies are urgently needed to promote its effective treatment. At Protheragen , we focus on developing novel therapeutics and building accurate animal models to accelerate preclinical studies of potential therapies for MHA.
May-Hegglin anomaly (MHA) is a rare genetically inherited disorder of platelets with an autosomal dominant pattern occurring due to mutations in the MYH9 gene. It is characterized by the triad of macrothrombocytopenia, pathognomonic Döhle-like cytoplasmic inclusions in leukocytes, and a variable tendency to bleed. MHA is in the spectrum of MYH9-related diseases, which MHA is mainly blood-related disorder. However, some genetic variants may show non-blood-related features such as hearing loss and glomerulopathy.
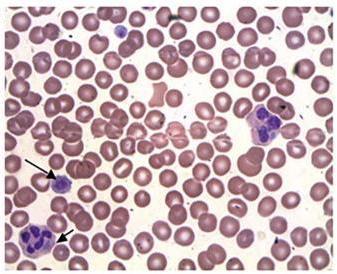 Fig.1 Giant platelets in the blood of a patient with May-Hegglin anomaly (MHA). (Kamath V, et al., 2016)
Fig.1 Giant platelets in the blood of a patient with May-Hegglin anomaly (MHA). (Kamath V, et al., 2016)
The pathogenesis of May-Hegglin anomaly (MHA) results from heterozygous mutations in the MYH9 gene which encodes non-muscle myosin heavy chain IIA NMMHC-IIA that alter the dynamics of the cytoskeleton in megakaryocytes and leukocytes. These mutations lead to disorganization of the actin-myosin networks which occurs during megakaryocyte maturation, thereby disrupting proplatelet formation and resulting in the generation of giant platelets.
 Fig.2 Key findings and clinical significance of MYH9-related disease from misdiagnosis to accurate diagnosis. (Cai L, et al., 2024)
Fig.2 Key findings and clinical significance of MYH9-related disease from misdiagnosis to accurate diagnosis. (Cai L, et al., 2024)
Supportive care remains the primary approach for treating May-Hegglin anomaly (MHA) because therapies targeting the underlying MYH9 mutation are unavailable. The main drug development pipeline is as follows:
| Drug Names | Mechanism of Action | Targets | Research Phase |
|---|---|---|---|
| Eltrombopag | Binds to and activates the thrombopoietin receptor (TPO-R) on megakaryocytes, stimulating platelet production. | Thrombopoietin receptor (c-Mpl) | Clinically used |
| DDAVP | Enhances platelet adhesion by increasing plasma von Willebrand factor (VWF) and factor VIII levels through endothelial release. | Vasopressin V2 receptors | Clinically used |
Disclaimer: Protheragen focuses on providing preclinical research services. This table is for information exchange purposes only. This table is not a treatment plan recommendation. For guidance on treatment options, please visit a regular hospital.
As your specialized research service provider, Protheragen strives to expand the scientific frontiers of rare bleeding disorders, particularly May-Hegglin anomaly (MHA). Our complete offerings comprise the development of diagnostics and therapeutics, precise disease modeling, and exhaustive preclinical validation.
MYH9 knockout models are constructed using either gene editing or classical homologous recombination.
Protheragen remains uncompromising in conducting an elaborate sequence of pharmacodynamics (PD), pharmacokinetics (PK) and toxicology studies for validating and refining therapies relating to May-Hegglin anomalies (MHA). For further information about our services or for quotations on related services, do not hesitate to reach out to us.
References
Protheragen's preclinical research services evaluate the pharmacodynamics, pharmacokinetics, and safety of drug candidates for rare blood disorders.
Myeloproliferative Disorders (MPDs)
Bone Marrow Failure Syndromes (BMFS)